


Technological innovations in genome sequencing, high-resolution imaging technologies and analysis tools have evolved with recent discovery in life sciences. On the research path to delivering cures, precision medicine and better healthcare are mountains of data pushed through bioinformatics workflow. Western Digital storage is there at every analysis, along the lifecycle of data, enabling new discoveries for genomics research, clinical research, pharma, and healthcare initiatives.
[Tweet “mountains of data are pushed through the #bioinformatics workflow. Here’s how it happens #genomics”]From intense parallel computation, to collaborative access or an long-lasting relative data at rest, here’s an overview of a typical bioinformatics workflow, and how innovations in flash and object storage can target and optimize genomics lifecycle regimes.
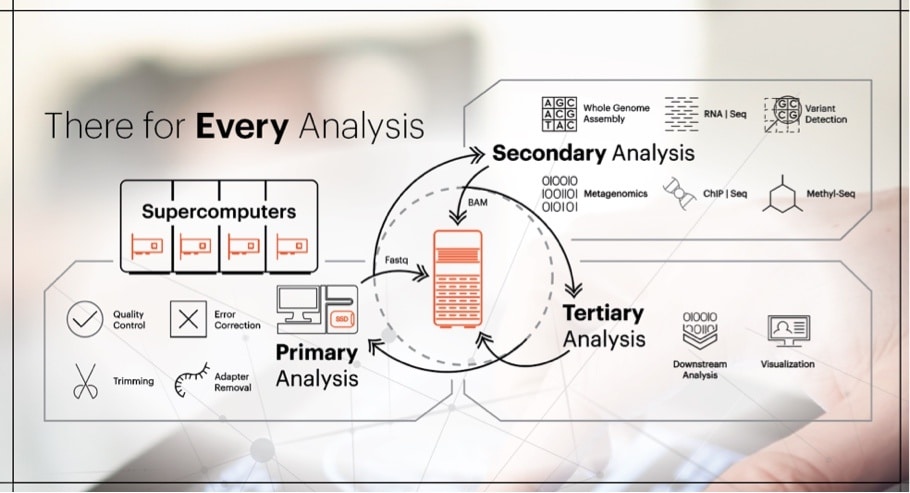
Primary Analysis
In the first stage of bioinformatics workflow data from instruments such as microscopes or sequencers is collected, often as multi-terabyte files[1]. Before the data is analyzed, the first step is to pre-process data. This includes quality control, trimming and filtering to clean the data in preparation for analysis. Once data is ready, it is ingested to supercomputers to generate DNA sequencing. This stage of the analysis employs high performance computing (HPC) by leveraging parallel file systems to distribute the workloads across HPC compute clusters. High performance computation can take advantage of the low latency of flash. Our HGST PCIe SSDs, with ultra-low consistent latency and extreme performance, support such clusters to help speed up analysis and can be used as overflow for data sets larger than physical memory, a bigger scratch space, and check pointing.
When the HPC cluster completes its tasks, outputs of this phase of analysis are FASTQ files, a format that contains both DNA sequence data and quality scores in a single file. Once created, FASTQ files are preserved for further analysis. As you can see in the diagram above, in a two-tier environment, FASTQ will be moved to efficient high-capacity storage such as our ActiveScaleTM object storage system.
Secondary Analysis
The secondary analysis, too, may have various pre-processing steps that are required. This includes operations such as combining files through analysis. Researchers then use FASTQ data to perform sequence alignment, a way of arranging DNA, RNA, or protein sequences to identify similarity and establish relationships between various sequences. The output of the alignment is stored in a Sequence Alignment Map (SAM) format, or in its compressed format, a Binary Alignment Map (BAM). Both forms are very large files sizes that require high-capacity storage. As you can see from the diagram above, following the secondary analysis, these BAM files return to be stored on the extremely durable, high-capacity ActiveScaleTM system.
Tertiary Analysis
Depending on the research focus, genome, protein, or molecular, various post-processing steps are performed in the tertiary analysis stage. The results from analysis like identifying variants, annotation, interpretation and data mining are often visualized for better understanding. These files, too, are preserved on high-capacity archives such as our object storage system depicted above.
Cyclical and Collaborative Analysis
Research is ever evolving, and research data is used recurrently and persistently. When new algorithms are released or genome sequencer manufacturer improves their software, researchers will often re-run their primary analysis, triggering secondary and tertiary re-runs.
Raw research data is also collaborative. In contrast to enterprise companies who safeguard their data in privacy, the life sciences community shares raw data to collaborate. Sharing FASTQ or BAM files provide the community with access to larger genomic pools and test beds. Hence, many researchers may access data to run secondary or tertiary analysis without running preliminary analysis themselves. Most bioinformatics environments will have a large number of BAM files from research universities, national genome banks and other sources.
Object Storage at the Heart of Every Step
NAS has been the primary storage tier for bioinformatics workflows. However, it is becoming a challenge for life sciences. Life sciences data is exploding, and NAS is a primary storage that’s costly and difficult to scale. Furthermore, at scale, NAS systems are hard to manage, and with hard drive capacity growing to 12 terabytes and beyond, RAID rebuild times can halt NAS to a crawl.
Object storage is a new alternative that contends well with data growth, long-term data preservation and random access of data. Its erasure coding algorithm is a far more efficient way to protect data than RAID at scale (our ActiveScale system delivers up to 17 nines of data durability and site-level fault tolerance in a multi-site configuration), and finding files is easy within a single global namespace across petabytes of storage. Object storage is cost efficient and often more cost effective than support for 4th or 5th year legacy NAS, and is a cloud-ready environment that simplifies collaboration internally or with other institutions.
Western Digital at Every Step of Bioinformatics Workflow
Western Digital solutions enable every step of bioinformatics workflow and help remove storage bottlenecks in genomic analysis. We help organizations quickly store, process, analyze and retain extreme amounts of data through innovative high-performance PCIe SSD solutions and high-capacity object storage systems. We offer unified storage that supports a diverse set of life sciences applications and workloads (including native protocol support for NFS, SMB, and Object) where performance, reliability, and availability of data are essential to the business.
Learn More
[1] http://events.linuxfoundation.org/sites/events/files/slides/Keynote_Sam%20Greenblatt_Wednesday.pdf